
Please set your exam date
Drug action: pharmaceutic, pharmacokinetic, and pharmacodynamic phases
Study Questions
Practice Exercise
The nurse anticipates that the health care provider will order which laboratory test for an older adult with renal dysfunction?
Explanation
Renal function assessment in older adults requires the most accurate measure of kidney filtration capacity. Creatinine clearance testing is the gold standard for estimating glomerular filtration rate (GFR) and identifying renal impairment. Since muscle mass decreases with age, serum creatinine alone may appear normal despite reduced kidney function, making creatinine clearance especially important.
Rationale for correct answer:
A. Creatinine clearance – This test measures the rate at which creatinine is cleared from the blood by the kidneys and closely reflects the GFR. In older adults with reduced muscle mass, it provides a more accurate indicator of renal function than serum creatinine alone.
Rationale for incorrect answers:
B. Glomerular filtration rate – While GFR is a key measure of kidney function, it is often estimated based on creatinine clearance rather than measured directly in routine practice. Direct creatinine clearance testing is more precise in this setting.
C. Urine specific gravity – This measures urine concentration and hydration status but does not accurately evaluate overall renal function or filtration capacity.
D. Urine pH – This indicates the acidity or alkalinity of urine, which can help in diagnosing some metabolic or urinary disorders, but it does not reflect kidney filtration efficiency.
Take-home points:
- Creatinine clearance is the most accurate test for assessing renal function in older adults.
- Serum creatinine may be misleading in elderly patients due to reduced muscle mass.
- Kidney filtration assessment is essential before prescribing drugs excreted by the kidneys.
A student nurse is studying the phases of drug action. Which statement by the student indicates to the nursing instructor that the student understands the pharmaceutic phase?
Explanation
The pharmaceutic phase refers to the initial stage of drug action for oral medications, where the drug is disintegrated and dissolved so it can be absorbed into body fluids and tissues. This phase is unique to oral dosage forms and precedes the pharmacokinetic and pharmacodynamic phases.
Rationale for correct answer:
D. “The pharmaceutic phase is the process by which the drug becomes available to body fluids and tissue.” – This is correct because it defines the phase as the breakdown and dissolution of the drug so it can enter circulation for absorption.
Rationale for incorrect answers:
A. “To achieve drug action, drugs are moved by four processes.” – This describes the pharmacokinetic phase, which involves absorption, distribution, metabolism, and excretion, not the pharmaceutic phase.
B. “For the drug to cross the biologic membrane, the drug becomes a solution.” – While dissolution into a solution is part of the pharmaceutic phase, this statement focuses only on one step and does not capture the full definition of the phase.
C. “In this phase, drugs are concentrated and a biologic or physiologic response occurs.” – This describes the pharmacodynamic phase, where the drug interacts with receptors to produce effects, not the pharmaceutic phase.
Take-home points:
- The pharmaceutic phase occurs only with oral medications.
- It involves disintegration and dissolution before absorption.
- It precedes the pharmacokinetic and pharmacodynamic phases.
A client is prescribed phenobarbital sodium (Luminal) for a seizure disorder. The medication has a long half-life of 4 days. Based on this half-life, the medication will most likely be prescribed
Explanation
Phenobarbital sodium has a long half-life of approximately 4 days, meaning it remains in the body for an extended period before being eliminated. Drugs with long half-lives generally require less frequent dosing to maintain therapeutic blood levels while avoiding drug accumulation and toxicity. This allows for once-daily administration in most cases, which can also improve medication adherence.
Rationale for correct answer:
A. Once a day – The long half-life means the drug stays in circulation for several days, so once-daily dosing is sufficient to maintain stable plasma concentrations and control seizures
Rationale for incorrect answers:
B. Twice a day – Unnecessary for a drug with such a prolonged half-life, as this could lead to excessive drug accumulation and an increased risk of sedation or toxicity.
C. Three times a day – Appropriate for short-acting medications but not for long-acting barbiturates like phenobarbital.
D. Four times a day – This frequent dosing is used for drugs with very short half-lives; with phenobarbital, it would cause dangerously high plasma drug levels and adverse effects.
Take-home points:
- Long half-life drugs require less frequent dosing to maintain therapeutic levels.
- Phenobarbital can be given once daily for seizure control due to its prolonged duration of action.
- More frequent dosing of long-acting drugs increases the risk of toxicity without improving efficacy.
A nurse educator is reviewing medication dosages and factors that influence medication metabolism with a group of nurses. Medication dosages may need to be decreased for which of the following reasons? Select all that apply
Explanation
Medication metabolism is primarily carried out in the liver, and any condition or interaction that reduces the liver’s ability to break down drugs can lead to increased drug levels in the body. In such cases, the dosage must be decreased to avoid toxicity. Additionally, when two drugs are metabolized through the same enzymatic pathway, they compete for metabolism, slowing the clearance of one or both medications.
Rationale for correct answers:
3. Liver failure – Reduced liver function decreases metabolism, causing drugs to stay in the system longer, requiring lower dosages to prevent toxicity.
5. Concurrent use of medication metabolized by the same pathway – Competition for the same metabolic enzymes slows drug clearance, increasing drug levels and requiring dosage reduction.
Rationale for incorrect answers:
1. Increased renal excretion – This speeds up drug elimination through the kidneys, potentially requiring an increased, not decreased, dosage to maintain therapeutic effects.
2. Increased medication-metabolizing enzymes – This accelerates metabolism, leading to lower drug levels; dosages may need to be increased rather than decreased.
4. Peripheral vascular disease – This may impair drug delivery to tissues but does not directly impact drug metabolism in the liver.
Take-home points:
- Liver failure reduces metabolism, increasing the risk of toxicity.
- Drug-drug metabolic competition can slow clearance and raise blood concentrations.
- Dosage adjustments are based on how metabolism and elimination are affected, not just the presence of disease.
A client with advanced liver disease is prescribed a drug with high first-pass metabolism. Which route is most appropriate to avoid reduced bioavailability?
Explanation
Drugs with high first-pass metabolism undergo extensive breakdown in the liver before reaching systemic circulation when given orally. In a client with advanced liver disease, this effect is even more pronounced, greatly reducing bioavailability. Administering the drug sublingually allows it to be absorbed directly into the systemic circulation via the oral mucosa, bypassing the liver initially and maintaining therapeutic levels.
Rationale for correct answer:
B. Sublingual – Absorption under the tongue delivers the drug directly into systemic circulation, bypassing the liver’s first-pass effect. This route is especially useful for drugs with high hepatic metabolism or in patients with compromised liver function. It allows for faster onset and more predictable plasma drug concentrations.
Rationale for incorrect answers:
A. Oral – Drugs taken orally are absorbed through the GI tract and pass through the liver via the portal circulation, undergoing extensive first-pass metabolism, which reduces bioavailability. This is not ideal for patients with significant liver impairment.
C. Rectal – While partial absorption from the lower rectum can bypass the liver, a significant portion of rectal blood flow still goes through the portal vein, meaning some first-pass metabolism still occurs.
D. Sustained-release oral tablet – Although it provides slow, steady drug release, it is still absorbed in the GI tract and subject to the first-pass effect, making it unsuitable for avoiding reduced bioavailability in this scenario.
Take-home points:
- Sublingual administration bypasses the liver and avoids first-pass metabolism.
- High first-pass metabolism drugs have reduced oral bioavailability, especially in liver disease.
- Drug route selection can significantly impact absorption, onset, and therapeutic effect.
A nurse should advise a client not to crush an enteric-coated tablet primarily because:
Explanation
Enteric-coated tablets are designed with a special coating that allows them to pass through the stomach intact and dissolve in the alkaline environment of the small intestine. Crushing the tablet destroys this coating, leading to premature drug release in the stomach, which can result in reduced effectiveness, irritation of the gastric mucosa, or altered absorption patterns.
Rationale for correct answer:
A. It may cause premature dissolution and alter absorption site – The coating protects the drug from stomach acid and protects the stomach from potential irritation. Crushing disrupts this process, causing drug release in the wrong part of the GI tract, altering its therapeutic effect and increasing the risk of gastric irritation.
Rationale for incorrect answers:
B. It reduces the drug’s potency, decreasing risk of toxicity – Crushing does not inherently reduce drug potency; instead, it can lead to faster absorption and possibly increased toxicity if the entire dose is released at once.
C. It ensures the drug is dissolved only in acidic pH environments – Enteric coatings are made to dissolve in alkaline, not acidic, environments. This statement reflects a misunderstanding of the drug’s design.
D. It eliminates the possibility of delayed absorption – While crushing can speed absorption, the primary concern is loss of targeted release and site-specific absorption, not just timing.
Take-home points:
- Enteric coating protects both the drug from stomach acid and the stomach from irritation.
- Crushing can cause premature drug release and change absorption location.
- Always check drug formulation before altering its form to avoid safety and efficacy issues.
A nurse is teaching a student nurse about bioavailability. Which of the following statements about bioavailability is most accurate?
Explanation
Bioavailability refers to the percentage of an administered drug that reaches the systemic circulation in its active form. For intravenous (IV) administration, the drug enters directly into the bloodstream without absorption barriers or first-pass metabolism, making its bioavailability 100%. This contrasts with oral drugs, which undergo variable absorption and hepatic metabolism before reaching systemic circulation.
Rationale for correct answer:
B. Intravenous drugs have 100% bioavailability – IV drugs bypass absorption barriers and the first-pass effect, delivering the entire administered dose into circulation, resulting in complete systemic availability.
Rationale for incorrect answers:
A. It refers only to the rate of GI drug dissolution – Bioavailability involves both the rate and extent of absorption into systemic circulation, not just GI dissolution.
C. Oral drugs generally have bioavailability close to 100% – Most oral drugs have less than 100% bioavailability due to incomplete absorption and first-pass metabolism in the liver.
D. The first-pass effect increases oral bioavailability – The first-pass effect decreases oral bioavailability because a portion of the drug is metabolized in the liver before reaching systemic circulation.
Take-home points:
- IV administration provides 100% bioavailability by directly entering the bloodstream.
- Oral bioavailability is reduced by absorption barriers and first-pass metabolism.
- Bioavailability measures both rate and extent of drug absorption into systemic circulation.
In a client with low albumin levels, what is the likely effect on a highly protein-bound drug?
Explanation
Albumin is the primary plasma protein that binds many drugs in the bloodstream. Highly protein-bound drugs rely on albumin for transport and controlled release. In a client with low albumin levels (e.g., due to liver disease, malnutrition, or chronic illness), fewer binding sites are available, resulting in more unbound or “free” drug in circulation
Rationale for correct answer:
B. Increased free drug levels and toxicity risk – With reduced protein binding, the free active form of the drug rises, enhancing therapeutic and potentially toxic effects. This is particularly important for drugs with narrow therapeutic ranges.
Rationale for incorrect answers:
A. Decreased drug effect due to more binding – Low albumin leads to less binding, not more. This would increase, not decrease, the drug’s effect.
C. Accelerated liver metabolism – Albumin levels do not directly influence liver metabolism. The liver processes drugs based on enzyme activity, not plasma protein levels.
D. Delayed renal elimination – While protein binding can affect filtration, low albumin primarily impacts drug distribution, not elimination rate.
Take-home points:
- Low albumin increases the free fraction of protein-bound drugs.
- Higher free drug levels raise the risk of toxicity, especially in drugs with narrow therapeutic indices.
- Monitoring plasma levels is crucial when administering highly protein-bound drugs in hypo-albuminemic clients.
Practice Exercise 2
The nurse is caring for a patient with congestive heart failure who is receiving digoxin (Digitek, Lanoxicaps, Lanoxin). The nurse plans to take which action when administering digoxin?
Explanation
Digoxin has a narrow therapeutic range (typically 0.5–2 ng/mL), meaning small changes in drug concentration can lead to toxicity or subtherapeutic effects. In patients with congestive heart failure, maintaining the drug within this range is essential for maximizing cardiac benefits while preventing adverse effects such as arrhythmias, visual disturbances, and gastrointestinal upset.
Rationale for correct answer:
D. Monitor the therapeutic range of digoxin – Monitoring serum levels within 0.5–2 ng/mL ensures the drug remains effective without causing toxicity, which is especially important given its narrow safety margin.
Rationale for incorrect answers:
A. Check the peak levels of digoxin – Peak levels are not routinely used for digoxin monitoring, as they do not reliably indicate toxicity risk due to the drug’s tissue binding and long distribution phase.
B. Check the trough levels of digoxin – While trough levels (just before the next dose) are more useful than peaks, the primary focus is on maintaining overall therapeutic serum concentration rather than monitoring troughs alone.
C. Monitor for tachyphylaxis – Tachyphylaxis refers to rapid drug tolerance development; digoxin does not typically cause this.
Take-home points:
- Digoxin requires close monitoring due to its narrow therapeutic index.
- The safe serum range is generally 0.5–2 ng/mL.
- Signs of toxicity include arrhythmias, visual changes (yellow/green halos), and GI upset.
When reviewing the client’s medication regimen, the nurse understands that the interval of drug dosage is related to what?
Explanation
The half-life of a drug is the time required for half of the drug concentration in the body to be eliminated. This pharmacokinetic property determines how often a medication should be administered to maintain therapeutic levels without causing toxicity. A drug with a short half-life requires more frequent dosing, whereas a drug with a long half-life can be given less often while still maintaining effective plasma concentrations.
Rationale for correct answer:
A. Half-life – The dosing interval is directly influenced by the drug’s half-life, ensuring a steady therapeutic level and avoiding fluctuations that could cause subtherapeutic effects or toxicity.
Rationale for incorrect answers:
B. Stimulation of receptors – This describes a mechanism of action, not the factor that determines dosing frequency.
C. Therapeutic index – While it indicates the safety margin of a drug, it does not directly dictate the dosing interval; instead, it guides safe dosing limits.
D. Trough level – Trough levels help determine if a drug remains in the therapeutic range before the next dose but are a monitoring tool, not the primary determinant of the dosing schedule.
Take-home points:
• Half-life is the key factor in determining dosing frequency.
• Short half-life drugs require more frequent administration than long half-life drugs.
• Understanding half-life helps balance drug effectiveness and safety.
Which nursing assessment(s) will the nurse include when developing a patient medication plan? Select all that apply
Explanation
A comprehensive nursing assessment for medication planning involves gathering information that ensures safe, effective, and individualized therapy. This includes checking drug levels, understanding protein-binding characteristics, and recognizing nonspecific side effects that may affect multiple body systems.
Rationale for correct answers:
A. Check peak and trough levels of drugs – Measuring these levels helps determine whether a drug is within its therapeutic range, avoiding subtherapeutic dosing and preventing toxicity.
B. Check the drug literature for the protein-binding percentage of a drug – Knowing a drug’s protein-binding percentage helps predict how much of it will be in the active, unbound form, especially important in patients with low albumin levels.
C. Identify side effects of drugs that are nonspecific – Nonspecific side effects affect multiple organ systems and may complicate patient response monitoring; identifying them helps with early detection and management.
Rationale for incorrect answers:
D. Evaluate the patient’s reaction to a drug – While essential for ongoing care, this occurs after administration and is part of evaluation, not the initial development of a medication plan.
E. To enhance absorption, advise the patient to crush an enteric-coated tablet before taking – This is unsafe; crushing destroys the protective coating, leading to premature drug release, altered absorption, and potential gastric irritation.
Take-home points:
- Peak and trough levels help maintain drugs within the therapeutic range.
- Protein-binding knowledge is vital for dosing decisions, especially in hypoalbuminemia.
- Identifying nonspecific side effects allows earlier recognition of adverse reactions.
A nurse is reviewing a client’s health record and notes a new prescription by the provider to verify the trough level of the client’s medication. Which of the following actions should the nurse take?
Explanation
Trough levels represent the lowest concentration of a drug in the bloodstream, measured just before the next scheduled dose. This value helps determine if the medication remains in the therapeutic range and guides dose adjustments to prevent toxicity or subtherapeutic effects.
Rationale for correct answer:
A. Have a blood specimen obtained immediately prior to the next dose of medication – Trough levels must be measured right before the next scheduled dose to reflect the lowest drug concentration in the body. This ensures accurate therapeutic monitoring.
Rationale for incorrect answers:
B. Verify that the client has been on the medication for 24-hr before ordering a blood specimen – Duration on the medication is not the determining factor; timing in relation to the next dose is critical.
C. Ask the client to provide a urine specimen after the next dose of medication – Trough levels are determined from blood samples, not urine, as urine levels do not reliably indicate circulating drug concentration.
D. Begin administering the medication, and obtain a blood specimen – Drawing blood after giving the dose would measure peak levels, not trough levels, and would not provide the intended monitoring information.
Take-home points:
- Trough levels are measured immediately before the next dose.
- They help prevent both toxicity and subtherapeutic dosing.
- Blood, not urine, is used for trough measurement.
Which of the following statements best describes pharmacodynamics?
Explanation
Pharmacodynamics focuses on the interaction between drugs and the body’s cells, tissues, and organ systems. It explains how drugs produce therapeutic effects, adverse effects, and toxic effects through mechanisms such as receptor binding, enzyme interactions, and non-specific cellular responses.
Rationale for correct answer:
B. What the drug does to the body, including biochemical and physiologic effects.
This is the definition of pharmacodynamics, which explains the drug’s mechanism of action and resulting effects on body systems.
Rationale for incorrect answers:
A. What the body does to the drug, including absorption, distribution, metabolism, and excretion.
This describes pharmacokinetics, not pharmacodynamics. Pharmacokinetics deals with the drug’s journey through the body rather than its effects.
C. The movement of a drug from its site of administration into the bloodstream.
This is absorption, a specific step within pharmacokinetics, not the broader concept of pharmacodynamics.
D. The dissolution of a drug into a solution in the GI tract.
This refers to the pharmaceutic phase, which occurs before absorption, and is unrelated to pharmacodynamic processes.
Take-home points:
- Pharmacodynamics = drug’s effect on the body’s systems and functions.
- Pharmacokinetics = body’s effect on the drug (ADME process).
- Knowing both helps predict drug response and guide safe medication administration.
A client receives a loading dose of an antibiotic. The nurse understands that the purpose of this is to:
Explanation
A loading dose is a larger initial dose of a medication given to quickly raise the drug’s plasma concentration to a therapeutic level. This is often used when the drug has a long half-life or when a rapid therapeutic effect is needed. Once the therapeutic concentration is achieved, the patient transitions to maintenance doses to sustain the desired effect.
Rationale for correct answer:
B. Rapidly achieve a therapeutic plasma concentration.
This is the purpose of a loading dose—to bypass the time it would take for the drug to accumulate gradually, ensuring the patient receives benefit sooner.
Rationale for incorrect answers:
A. Maintain steady-state drug levels over time.
This is the role of maintenance dosing, not a loading dose. Steady-state levels are achieved gradually after repeated doses.
C. Increase the drug’s potency.
Potency is an inherent property of the drug and is not affected by dosage size. A loading dose changes plasma concentration, not potency.
D. Reduce the likelihood of drug toxicity.
A loading dose may actually increase the risk of toxicity if not calculated correctly, since it involves giving a higher initial dose.
Take-home points:
- Loading dose = rapid therapeutic concentration.
- Used when drug has long half-life or when urgent treatment is needed.
- Followed by maintenance doses to keep plasma levels steady.
When comparing morphine and tramadol for analgesia, the fact that morphine produces greater maximum pain relief than tramadol refers to which of the following:
Explanation
When comparing drugs, efficacy refers to the maximum effect a drug can produce, regardless of dose. If one drug provides greater maximum therapeutic benefit than another—such as morphine producing stronger pain relief than tramadol—this difference reflects efficacy, not potency or absorption characteristics.
Rationale for correct answer:
B. Efficacy
Efficacy describes the drug’s ability to produce the greatest possible therapeutic effect. Morphine’s ability to produce more powerful pain relief than tramadol demonstrates greater efficacy.
Rationale for incorrect answers:
A. Potency
Potency is the amount of a drug needed to produce a given effect. A drug can be more potent without being more effective; for example, it may require a smaller dose to achieve the same effect, but the maximum effect may still be lower.
C. Onset
Onset is the time it takes for a drug’s effect to begin after administration. This does not address differences in the maximum possible relief.
D. Bioavailability
Bioavailability is the fraction of the administered dose that reaches the systemic circulation. While it can influence the amount of drug available, it does not determine the drug’s maximum therapeutic effect.
Take-home points:
- Efficacy = maximum possible therapeutic effect.
- Potency = dose required to achieve a given effect.,.,
- Higher efficacy means greater therapeutic benefit at the drug’s peak effect.
Comprehensive Questions
A client has an order for a tetracycline antibiotic and has been instructed to avoid taking the medication with foods, beverages, or drugs that contain calcium, iron, or magnesium. The client takes the antibiotic along with a daily multivitamin, not realizing that the vitamin contains iron. What effect may this have on the tetracycline?
Explanation
Tetracycline antibiotics are broad-spectrum medications used to treat various bacterial infections, including acne, respiratory infections, and certain sexually transmitted diseases. A critical consideration is that iron, calcium, magnesium, and antacids can impair tetracycline absorption by forming insoluble complexes. To maximize effectiveness, clients should be instructed to avoid taking tetracycline with iron supplements, dairy products, or antacids, allowing at least 1 to 2 hours between doses.
Rationale for correct answer:
A. Impaired absorption:
Iron in the multivitamin binds with tetracycline in the gastrointestinal tract, forming insoluble complexes that reduce the drug's absorption and effectiveness. This can lead to subtherapeutic antibiotic levels and treatment failure.
Rationale for incorrect answers:
B. Increased distribution:
Distribution refers to the movement of the drug into tissues and is not significantly influenced by the presence of iron. The primary concern with iron is its interaction in the gut, not in systemic circulation.
C. Decreased metabolism:
Iron does not impact liver enzymes that metabolize tetracycline. Tetracycline metabolism is more influenced by hepatic enzyme activity, which is not altered by iron supplementation.
D. Impaired excretion:
Tetracycline is excreted primarily by the kidneys, and iron does not interfere with renal elimination of the drug. The interaction occurs before the drug is absorbed, not during excretion.
- Avoid giving tetracyclines with iron, calcium, or magnesium—they impair drug absorption.
- Separate tetracycline from multivitamins, antacids, or dairy by 1–2 hours.
- Reduced absorption lowers drug effectiveness, risking treatment failure.
A client has a malignant brain tumor. What pharmacokinetic phase may be affected by the presence of the tumor?
Explanation
A malignant brain tumor is a rapidly growing mass of abnormal cells within the brain that can disrupt normal neurological function. These tumors may compromise the blood-brain barrier (BBB) or alter cerebral blood flow, which significantly impacts drug behavior in the body. In such cases, the distribution phase of pharmacokinetics may be affected, as the tumor can hinder or alter the transport of drugs to brain tissue, especially medications that rely on crossing the BBB for therapeutic effect.
Rationale for correct answer:
B. Distribution:
A malignant brain tumor can alter the blood-brain barrier, making it more or less permeable, which affects how drugs are distributed into the brain. Tumors may also disrupt local blood flow, impacting the amount of drug reaching the brain tissue.
Rationale for incorrect answers:
A. Absorption:
Absorption refers to the movement of a drug from the site of administration into the bloodstream. A brain tumor does not typically interfere with this phase unless it affects the gastrointestinal system, which is unlikely.
C. Metabolism:
Drug metabolism primarily occurs in the liver. While cancer may indirectly affect liver function, a brain tumor alone does not typically impair drug metabolism unless it has metastasized to hepatic tissue.
D. Excretion:
Excretion involves the elimination of drugs through the kidneys or bile. A brain tumor does not directly impact renal or biliary function, so this pharmacokinetic phase remains mostly unaffected.
Take-home points:
- Brain tumors can alter drug distribution due to changes in the blood-brain barrier.
- Distribution phase is critical when treating CNS conditions.
- Always consider anatomical or pathological barriers in drug therapy planning.
A client with cirrhosis of the liver exhibits decreased metabolic activity. This will require what possible changes? Select all that apply.
Explanation
A client with cirrhosis of the liver often exhibits decreased metabolic activity due to impaired liver function. The liver is a primary site for drug metabolism, and compromised hepatic function can reduce the breakdown of medications, leading to increased drug levels in the bloodstream. This situation may necessitate a reduction in drug dosage to prevent accumulation, a change in the timing of medication administration to allow more time for clearance, and more frequent monitoring for adverse drug effects to ensure safety and therapeutic effectiveness.
Rationale for correct answers:
A. A reduction in the dosage of the drugs:
Liver cirrhosis impairs drug metabolism, leading to slower clearance. Lower doses may be necessary to prevent accumulation and toxicity.
E. More frequent monitoring for adverse drug effects:
Impaired metabolism raises the risk of drug accumulation. Close monitoring helps detect early signs of toxicity and allows timely adjustments.
Rationale for incorrect answers:
B. A change in the timing of medication administration:
A change in the timing of administration may still include a drug dosage that is too great for the liver to metabolize and the dosage will still not have changed.
C. An increased dose of prescribed drugs:
Increasing the dose would likely lead to drug accumulation in clients with reduced liver function and could increase the risk of serious side effects or toxicity.
D. Giving all prescribed drugs by intramuscular injection:
The route of administration doesn't resolve the issue of impaired hepatic metabolism. IM injections may also be less desirable in cirrhotic patients due to bleeding risks.
Take-home points:
- Liver cirrhosis reduces drug metabolism, requiring dose adjustments.
- Monitoring is crucial to prevent toxicity.
- Avoid increasing doses or relying on IM routes unnecessarily.
The client requires a drug that is known to be completely metabolized by the first-pass effect. What change will be needed when this drug is administered?
Explanation
When a drug is known to undergo complete metabolism by the first-pass effect, it means that the liver inactivates most or all of the drug before it reaches systemic circulation when taken orally. To achieve the desired therapeutic effect, the drug must be administered through a non-oral route, such as parenterally (e.g., intravenous or intramuscular). This bypasses the hepatic first-pass metabolism and allows the drug to reach its target tissues effectively.
Rationale for correct answer:
D. The drug must be given by a nonoral route such as parenterally:
Giving the drug by injection (IV, IM, subQ) bypasses the liver’s first-pass metabolism, ensuring more of the active drug reaches systemic circulation.
Rationale for incorrect answers:
A. The drug must be given more frequently:
Increasing frequency doesn't overcome the first-pass effect. The liver will still inactivate much of the drug before it reaches systemic circulation when given orally.
B. The drug must be given in higher doses:
While higher doses may offset some loss, this approach risks hepatotoxicity or side effects without ensuring sufficient therapeutic levels in the bloodstream.
C. The drug must be given in a lipid-soluble form:
Lipid solubility affects absorption, not hepatic metabolism. A lipid-soluble form would still undergo first-pass metabolism if given orally.
Take-home points:
- First-pass effect refers to drug metabolism in the liver before reaching systemic circulation.
- Bypassing the liver via nonoral routes prevents significant drug inactivation.
- Route of administration is critical for drugs with high first-pass metabolism.
A client who is in renal failure may have a diminished capacity to excrete medications. The nurse must assess the client more frequently for what development?
Explanation
In patients with renal failure, the kidneys lose their ability to effectively eliminate medications from the body, leading to drug accumulation in the bloodstream. As a result, the patient faces an increased risk for drug toxicity, especially with drugs that are primarily excreted through the kidneys. The nurse must closely monitor for signs of adverse effects and report any symptoms that may indicate toxicity.
Rationale for correct answer:
C. Increased risk for drug toxicity:
Impaired renal excretion causes drugs to accumulate in the body, especially those eliminated by the kidneys. This accumulation raises plasma drug levels and the potential for toxicity.
Rationale for incorrect answers:
A. Increased risk of allergy:
Allergic reactions are immune responses and not directly related to impaired renal function. Renal failure does not inherently increase the likelihood of developing a drug allergy.
B. Decreased therapeutic drug effects:
Renal failure can cause drug accumulation, leading to excessive rather than diminished effects. Therefore, therapeutic levels may actually increase, not decrease.
D. Increased absorption of the drug from the intestines:
Renal failure does not directly affect gastrointestinal absorption. Drug absorption primarily depends on GI factors and not on kidney function.
Take-home points:
- Renal failure impairs drug excretion, leading to drug accumulation.
- Clients with impaired renal function are at higher risk for drug toxicity.
- Dose adjustments and frequent monitoring are essential in renal-compromised patients.
What is the rationale for the administration of a loading dose of a drug?
Explanation
When immediate drug effects are needed, a loading dose is often prescribed to quickly achieve therapeutic plasma concentrations. This approach is particularly useful for medications with long half-lives, where waiting for steady-state levels through regular dosing would take too long. By giving a larger initial dose, the drug reaches the plateau level more rapidly, allowing for faster clinical benefits.
Rationale for correct answer:
D. It more rapidly builds plasma drug levels to a plateau level:
A loading dose is a higher initial dose that helps quickly achieve therapeutic drug levels in the plasma, which is especially useful for drugs with long half-lives or when an immediate effect is desired.
Rationale for incorrect answers:
A. It decreases the number of doses that must be given:
A loading dose does not decrease the total number of doses required over time. It is used to initiate treatment rapidly but does not eliminate the need for maintenance doses afterward.
B. It results in lower dosages being required to achieve therapeutic effects:
The purpose of a loading dose is not to reduce future doses, but to quickly raise the drug concentration to a therapeutic level. Maintenance doses typically follow to keep the drug within the therapeutic range.
C. It decreases the risk of drug toxicity:
A loading dose can actually increase the risk of toxicity if not calculated correctly, especially with drugs that have narrow therapeutic indices. Careful dosing and monitoring are needed.
Take-home points:
- Loading doses rapidly achieve therapeutic drug levels.
- They are especially useful for drugs with long half-lives.
- Maintenance doses follow to keep plasma levels steady.
A client experiences profound drowsiness when a stimulant drug is given. This is an unusual reaction for this drug, a reaction that has not been associated with that particular drug. What is the term for this type of drug reaction?
Explanation
When a client has an unexpected or atypical response to a medication—such as profound drowsiness from a stimulant—this is referred to as an idiosyncratic reaction. These responses are not related to the drug’s known pharmacologic action and often result from genetic differences in drug metabolism or immune response. Such reactions require close monitoring and prompt reporting to ensure patient safety.
Rationale for correct answer:
B. Idiosyncratic reaction:
An idiosyncratic reaction is an uncommon and unpredictable response to a drug that occurs due to genetic differences or unknown mechanisms. Profound drowsiness from a stimulant, which typically causes alertness, is an example of this type of unusual response.
Rationale for incorrect answers:
A. Allergic reaction:
An allergic reaction is an immune-mediated response to a drug, often involving symptoms like rash, itching, swelling, or anaphylaxis. It does not explain an unexpected central nervous system effect like drowsiness from a stimulant.
C. Enzyme-specific reaction:
Enzyme-specific reactions refer to drug metabolism influenced by certain enzymes, like CYP450. While enzyme activity can alter drug levels, it doesn't explain an atypical response not previously linked to the drug.
D. Unaltered reaction:
This term is not a recognized classification of drug reactions. It does not apply to the scenario of an unusual or paradoxical drug effect.
Take-home points:
- Idiosyncratic reactions are rare, unpredictable, and not dose-dependent.
- They differ from allergic reactions, which involve the immune system.
- Genetic variability often plays a role in idiosyncratic responses.
The provider has ordered atropine, a drug that will prevent the client’s own chemical, acetylcholine, from causing parasympathetic effects. What type of drug would atropine be considered?
Explanation
Atropine is a medication that blocks the action of acetylcholine, a neurotransmitter responsible for parasympathetic nervous system responses such as slowed heart rate and increased glandular secretions. Because it binds to receptors without activating them, atropine is classified as an antagonist. Understanding this helps nurses anticipate its effects on heart rate, pupil size, and secretions during care.
Rationale for correct answer:
A. An antagonist:
An antagonist is a drug that binds to receptors but blocks or inhibits the action of endogenous chemicals. Atropine works by blocking acetylcholine at muscarinic receptors, thereby preventing parasympathetic responses, making it a classic example of an antagonist.
Rationale for incorrect answers:
B. A partial agonist:
A partial agonist activates a receptor but produces a weaker response than a full agonist. Atropine does not stimulate the receptor at all; it blocks it, so this classification does not apply.
C. An agonist:
An agonist binds to a receptor and mimics the action of the body’s own substances. Since atropine inhibits rather than stimulates receptor activity, it is not considered an agonist.
D. A protagonist:
"Protagonist" is not a pharmacological term. It typically refers to the main character in a narrative, not a drug action classification.
Take-home points:
- Antagonists block the effects of neurotransmitters like acetylcholine.
- Atropine is a classic parasympathetic antagonist used to reduce secretions or increase heart rate.
- Recognize the difference between agonists (activate) and antagonists (block) in drug mechanisms.
What is the term used to describe the magnitude of maximal response that can be produced from a particular drug?
Explanation
When evaluating drug therapy, one key concept is efficacy, which refers to the maximum therapeutic response a drug can produce, regardless of dose. A drug with high efficacy is capable of producing strong clinical effects, even if it’s not the most potent. Nurses must understand efficacy to choose or evaluate the best drug for achieving desired treatment outcomes.
Rationale for correct answer:
A. Efficacy:
Efficacy refers to the maximum effect a drug can produce, regardless of dose. It describes how well the drug works once it binds to the receptor and elicits a response.
Rationale for incorrect answers:
B. Toxicity:
Toxicity relates to the degree of harmful effects a drug can cause at high doses. It is not related to the drug's ability to produce a therapeutic effect.
C. Potency:
Potency refers to the amount of drug needed to produce a given effect. A drug can be highly potent but have low efficacy if it doesn’t produce a strong maximal response.
D. Comparability:
Comparability is not a formal pharmacological term used to describe a drug’s effectiveness or dose-response characteristics. It is more relevant in general or regulatory contexts.
Take-home points:
- Efficacy is about how effective a drug is at producing a maximal response.
- A drug with higher efficacy is often preferred over one with higher potency if it provides a better therapeutic outcome.
- Potency ≠ Efficacy — they describe different aspects of drug action.
The nurse looks up butorphanol (Stadol) in a drug reference guide prior to administering the drug and notes that it is a partial agonist. What does this term tell the nurse about the drug?
Explanation
When reviewing drug classifications, nurses may encounter partial agonists, such as butorphanol (Stadol). These drugs bind to receptors like full agonists but produce a weaker or less efficacious response. Understanding this helps the nurse anticipate moderate therapeutic effects and potentially fewer side effects, making partial agonists useful in cases where full receptor activation is not necessary.
Rationale for correct answer:
D. It is a drug that produces a weaker, or less efficacious, response than an agonist drug:
A partial agonist activates the receptor but produces a submaximal response compared to a full agonist. This makes it useful in situations where a milder effect is desirable, such as with butorphanol.
Rationale for incorrect answers:
A. It is a drug that produces the same type of response as the endogenous substance:
This describes a full agonist, which binds to receptors and produces a maximal response similar to the body's natural chemical.
B. It is a drug that will occupy a receptor and prevent the endogenous chemical from acting:
This defines an antagonist, which blocks receptor activation and prevents any response.
C. It is a drug that causes unpredictable and unexplained drug reactions:
This describes an idiosyncratic reaction, not a partial agonist. Idiosyncratic reactions are rare and unrelated to known drug mechanisms.
Take-home points:
- A partial agonist binds to receptors but produces submaximal effects.
- Partial agonists like butorphanol are often used for moderate pain and have less abuse potential.
- Understanding receptor action (agonist vs partial agonist vs antagonist) is key in drug classification and predicting effects.
The nurse reads that the drug to be given to the client, has a “narrow therapeutic index.” The nurse knows that this means that the drug has what properties?
Explanation
Drugs with a narrow therapeutic index (NTI) require careful monitoring because they have a small margin between therapeutic and toxic levels. Even slight increases in dosage can lead to serious adverse or toxic effects. Nurses must be vigilant with dose calculations, monitoring levels, and assessing for toxicity to ensure safe administration.
Rationale for correct answer:
B. It has a narrow safety margin and even a small increase in dose may produce adverse or toxic effects:
Drugs with a narrow therapeutic index require precise dosing because minor changes in blood concentration can lead to toxicity or subtherapeutic effects, making careful monitoring essential.
Rationale for incorrect answers:
A. It has a narrow range of effectiveness and may not give this client the desired therapeutic results:
A narrow therapeutic index does not refer to a drug’s overall effectiveness but rather to the small margin between therapeutic and toxic levels, requiring close monitoring for safety.
C. It has a narrow range of conditions or diseases that the drug will be expected to treat successfully:
This describes a drug’s therapeutic application or spectrum, not its therapeutic index. A drug can have a wide or narrow spectrum regardless of its therapeutic index.
D. It has a narrow segment of the population for whom the drug will work as desired:
This reflects variability in individual response to the drug rather than the concept of a therapeutic index, which concerns dosage safety rather than population specificity.
Take-home points:
- A narrow therapeutic index drug has a tight margin between safe and toxic levels.
- Such drugs require frequent serum level monitoring to ensure patient safety.
- Nurses should be vigilant for signs of toxicity and ensure accurate dosing and timing
Which components of pharmacokinetics does the nurse need to understand before administering a drug? Select all that apply.
Explanation
Pharmacokinetics involves how the body absorbs, distributes, metabolizes, and excretes drugs. In clients with kidney disease, reduced protein-binding capacity can lead to more free (active) drug in circulation, increasing the risk of toxicity. Additionally, decreased metabolic rates slow drug breakdown, allowing drug accumulation, which can further heighten the potential for adverse effects. Close monitoring is essential in these patients to adjust dosages appropriately.
Rationale for correct answers:
C. Clients with kidney disease may have fewer protein-binding sites and are at risk for drug toxicity:
Kidney disease can reduce albumin levels, decreasing protein binding. More free (active) drug remains in circulation, increasing the risk of toxicity.
E. When the drug metabolism rate is decreased, excess drug accumulation can occur, which can cause toxicity:
If the liver or enzyme systems metabolize drugs more slowly, the drug can accumulate in the body over time, increasing the risk of adverse effects or toxicity.
Rationale for incorrect answers:
A. Drugs with a smaller volume of drug distribution have a longer half-life:
A smaller volume of distribution typically means the drug remains more in the bloodstream and is eliminated faster, resulting in a shorter half-life, not a longer one.
B. Oral drugs are dissolved through the process of pinocytosis:
Most oral drugs are absorbed via passive diffusion, active transport, or facilitated diffusion. Pinocytosis is rare and usually associated with large molecules like vitamins, not common oral drugs.
D. Rapid absorption decreases the bioavailability of the drug:
Rapid absorption generally increases bioavailability unless the drug is extensively metabolized first-pass. Decreased bioavailability is more associated with poor absorption or extensive first-pass metabolism.
Take-home points:
- Impaired kidney or liver function can significantly alter pharmacokinetics, especially metabolism and excretion.
- Reduced protein-binding and slower metabolism increase risk of drug toxicity.
- Nurses must assess for organ impairment and monitor closely when administering medications.
The nurse will question the health care provider if a drug with a half-life (t 1/2 ) of more than 24 hours is ordered to be given more than how often?
Explanation
When a drug has a half-life greater than 24 hours, it remains active in the body for an extended period. Administering such a drug more than once daily can lead to drug accumulation and toxicity. The nurse must understand half-life principles and should question the provider if the dosing frequency appears too high based on the drug's pharmacokinetics.
Rationale for correct answer:
A. Once daily:
A drug with a half-life over 24 hours remains in the body for a prolonged period, so giving it more than once daily may cause accumulation and toxicity. The nurse should question dosing more frequently than once a day.
Rationale for incorrect answers:
B. Every other day:
This frequency may be appropriate for drugs with a long half-life, as it allows time for partial elimination and helps maintain therapeutic levels without reaching toxic concentrations.
C. Twice weekly:
This interval can be suitable for certain drugs with very long half-lives. It helps maintain stable blood levels while minimizing the risk of accumulation.
D. Once weekly:
Some drugs with very long half-lives (e.g., bisphosphonates) are specifically designed for weekly dosing. This frequency supports compliance and safety in those cases.
Take-home points:
- Half-life (t½) affects how often a drug should be given.
- Drugs with long half-lives require less frequent dosing to avoid toxicity.
- Nurses must question orders that do not match pharmacokinetic principles.
The nurse is explaining drug action to a nursing student. Which statement made by the nurse is correct?
Explanation
Drugs in the bloodstream often bind to plasma proteins like albumin. Only the unbound (free) portion of the drug is pharmacologically active and able to exert therapeutic effects. Understanding protein binding helps the nurse anticipate drug efficacy and potential toxicity, especially in patients with low albumin levels or altered protein-binding capacity.
Rationale for correct answer:
B. “A drug not bound to protein is an active drug.”
Only the free (unbound) portion of a drug can exert a pharmacological effect. Drugs bound to plasma proteins like albumin are inactive until released.
Rationale for incorrect answers:
A. “Water-soluble and ionized drugs are quickly absorbed.”
Water-soluble and ionized drugs tend to have slower absorption because they do not easily cross lipid-rich cell membranes. Lipid-soluble, non-ionized drugs are absorbed more rapidly.
C. “Most receptors are found under the cell membrane.”
Most drug receptors are located on the surface of the cell membrane, not beneath it. They interact with circulating substances to produce effects.
D. “Toxic effects can result if the trough level is low.”
Toxicity typically occurs when drug levels are too high. A low trough level might lead to subtherapeutic effects, not toxicity.
Take-home points:
- Only free (unbound) drugs are active and can cause a pharmacologic response.
- Protein-binding affects drug distribution, action, and elimination.
- Nurses must understand basic drug dynamics to monitor safety and effectiveness.
The nurse plans to advise the client to avoid which food(s) before ingesting an enteric-coated medication? Select all that apply.
Explanation
Enteric-coated medications are designed to bypass the stomach and dissolve in the intestines to prevent gastric irritation or degradation. Consuming high-fat foods such as ice cream or fried chicken before taking these drugs can delay gastric emptying and interfere with proper dissolution and absorption, potentially reducing the medication's effectiveness.
Rationale for correct answers:
D. Ice cream:
Ice cream is high in fat and may delay gastric emptying, which can prevent the enteric coating from dissolving in the intestines as intended. This can alter drug effectiveness.
E. Fried chicken:
Fried chicken is high in fat, which slows gastric emptying. This can delay the medication reaching the alkaline environment of the small intestine, where the enteric coating is meant to dissolve.
Rationale for incorrect answers:
A. Bananas:
Bananas are low in fat and fiber and unlikely to interfere with the dissolution or absorption of an enteric-coated medication. They are generally safe to consume with medications.
B. Baked lamb chops:
Though a protein-rich food, baked lamb chops are not high in fat or acidic content that would impact enteric-coated medication. They typically do not delay gastric emptying significantly.
C. Broiled fish:
Broiled fish is low in fat and easy to digest. It does not pose a concern for altering the timing or breakdown of enteric-coated medications in the GI tract.
Take-home points:
- Enteric-coated drugs are designed to dissolve in the intestine, not the stomach.
- High-fat foods like ice cream and fried foods can disrupt the protective coating, leading to early drug release and reduced therapeutic effect.
- Educate patients to avoid high-fat meals around the time of taking enteric-coated medications.
Exams on Drug action: pharmaceutic, pharmacokinetic, and pharmacodynamic phases
Custom Exams
Login to Create a Quiz
Click here to loginLessons
Notes Highlighting is available once you sign in. Login Here.
Objectives
- Differentiate the three phases of drug action (pharmaceutic, pharmacokinetic, pharmacodynamic) and their clinical relevance.
- Explain how drug absorption is influenced by route, formulation, and physiological factors.
- Understand the role of protein binding and distribution in drug effectiveness and toxicity.
- Describe drug metabolism, excretion, and the impact of liver or kidney dysfunction.
- Apply receptor theory to explain drug actions, including agonists and antagonists.
- Interpret dose-response relationships, therapeutic index, and drug safety margins.
- Identify onset, peak, and duration of drug action and their importance in dosing.
- Recognize adverse effects, tolerance, pharmacogenetics, and individual variability in drug response.
Introduction
Understanding drug action involves three fundamental and sequential phases: the pharmaceutic, pharmacokinetic, and pharmacodynamic phases.
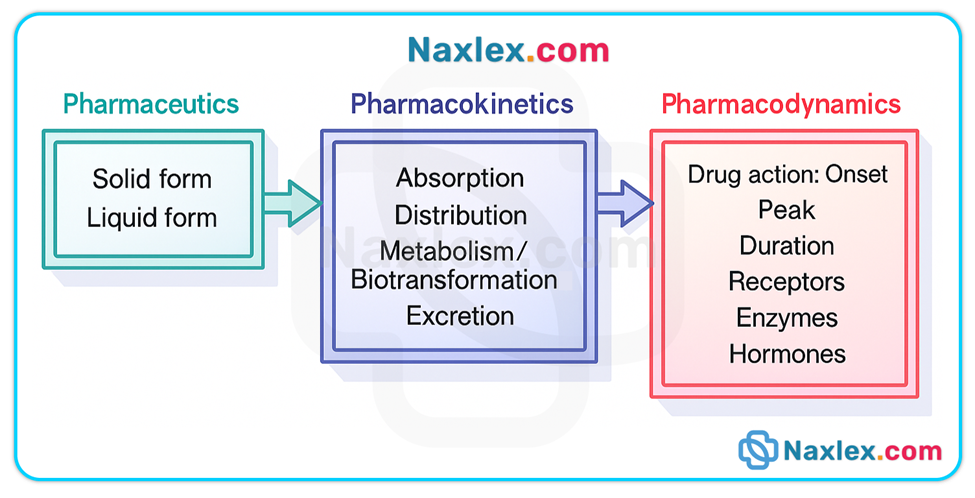
These phases describe how a drug is transformed from a dosage form into an active entity in solution, how the body handles that drug (ADME—absorption, distribution, metabolism, excretion), and how the drug produces a biologic or physiologic response.
The pharmaceutic phase applies only to oral solid dosage forms (tablets, capsules) and includes disintegration and dissolution so the drug can cross biologic membranes. Parenteral, sublingual, transdermal, inhaled, and many topical routes bypass this phase.
The pharmacokinetic phase (ADME) determines onset, intensity, duration, and elimination of drug effect.
The pharmacodynamic phase explains how a drug produces a response at receptor or effector sites, and why therapeutic and adverse effects occur.
Clinical relevance / pathophysiology links: organ dysfunction (GI disease, liver disease, kidney disease), age-related physiologic changes, and genetic differences alter one or more phases and therefore change dosing, onset, duration, and toxicity risk.
Understanding each phase allows safe medication administration, appropriate monitoring, patient education, and early recognition of altered drug responses.
Pharmaceutic phase
(Only for oral solid dosage forms — tablets and capsules)
Definition & overview
• The pharmaceutic phase is the process by which a solid oral dosage form disintegrates into smaller particles and then dissolves (dissolution) into GI fluids so the drug is in solution and available for absorption across biologic membranes. Liquid forms (solutions, syrups) are already in solution and bypass disintegration.
• Approximately 80% of drugs are taken by mouth; therefore pharmaceutic-phase processes are highly relevant to clinical nursing.
Key processes
• Disintegration — mechanical/chemical breakdown of a tablet/capsule into smaller particles.
• Dissolution — the subsequent dissolving of those particles into GI fluid to form a solution that can cross membranes.
Formulation factors that influence the pharmaceutic phase
• Excipients / fillers / binders / disintegrants — inert ingredients added to achieve proper size, shape, stability, or to enhance dissolution (e.g., some salts increase solubility).
• Particle size — smaller particles have greater surface area and dissolve faster.
• Coatings / special formulations — enteric-coated tablets resist stomach acid and dissolve in the more alkaline small intestine; sustained- / extended-release formulations release drug slowly over time. Crushing or splitting these preparations alters intended release and can cause toxicity or therapeutic failure.
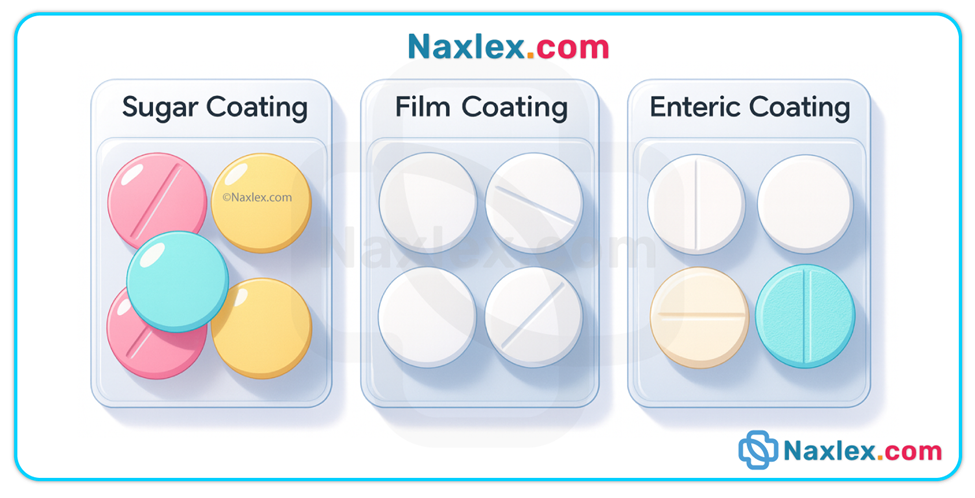
• Liquid vs solid forms — liquids are generally absorbed faster because they bypass disintegration and dissolution.
Physiologic & pathophysiologic influences
• Gastric pH: the stomach’s acidity (pH 1–2) promotes disintegration/dissolution of many drugs; alkaline environments may ionize some drugs and reduce membrane crossing. Infants and older adults often have less gastric acidity (hypochlorhydria) → slower dissolution and absorption of drugs normally absorbed in the stomach.
• Gastric motility & emptying time: delayed emptying (gastroparesis, opioids, postoperative ileus) prolongs contact of drug with stomach, delaying arrival to small intestine (where many drugs are absorbed). Rapid emptying shortens contact time.
• Food & fluid: food can delay, enhance, or reduce dissolution depending on the drug (e.g., fat-rich meals slow gastric emptying; some drugs cause gastric irritation and require administration with food).
• Surgical changes: resections (partial gastrectomy, bowel resection) reduce surface area, alter pH, and substantially change dissolution/absorption dynamics.
Examples & mechanistic notes
• Penicillin salts: penicillin G is acid-labile and poorly absorbed in gastric acid; forming sodium or potassium salts increases absorbability and mitigates gastric destruction.
• Enteric-coated tablets: can remain in stomach for prolonged periods; onset may be delayed if gastric emptying is slow.
Nursing insights
• Determine whether a tablet/capsule may be crushed or split — consult drug references or pharmacy before altering dosage forms. Never crush enteric-coated, sustained-release, or otherwise modified-release products unless explicitly allowed.
• Assess swallowing safety; consider alternative formulations (liquid, sublingual, transdermal, parenteral) for patients with dysphagia.
• Take medication administration times and meal instructions seriously — provide education about why certain drugs are taken with or without food.
• Be alert for patient factors that delay dissolution (PPI therapy, antacids, bariatric surgery, gastroparesis) and consider route changes when indicated.
Pharmacokinetic phase
(What the body does to the drug — ADME)
Overview
Pharmacokinetics consists of four interrelated processes: Absorption, Distribution, Metabolism (biotransformation), and Excretion (elimination). These processes determine the concentration-time profile of a drug in the body and therefore its clinical effects.
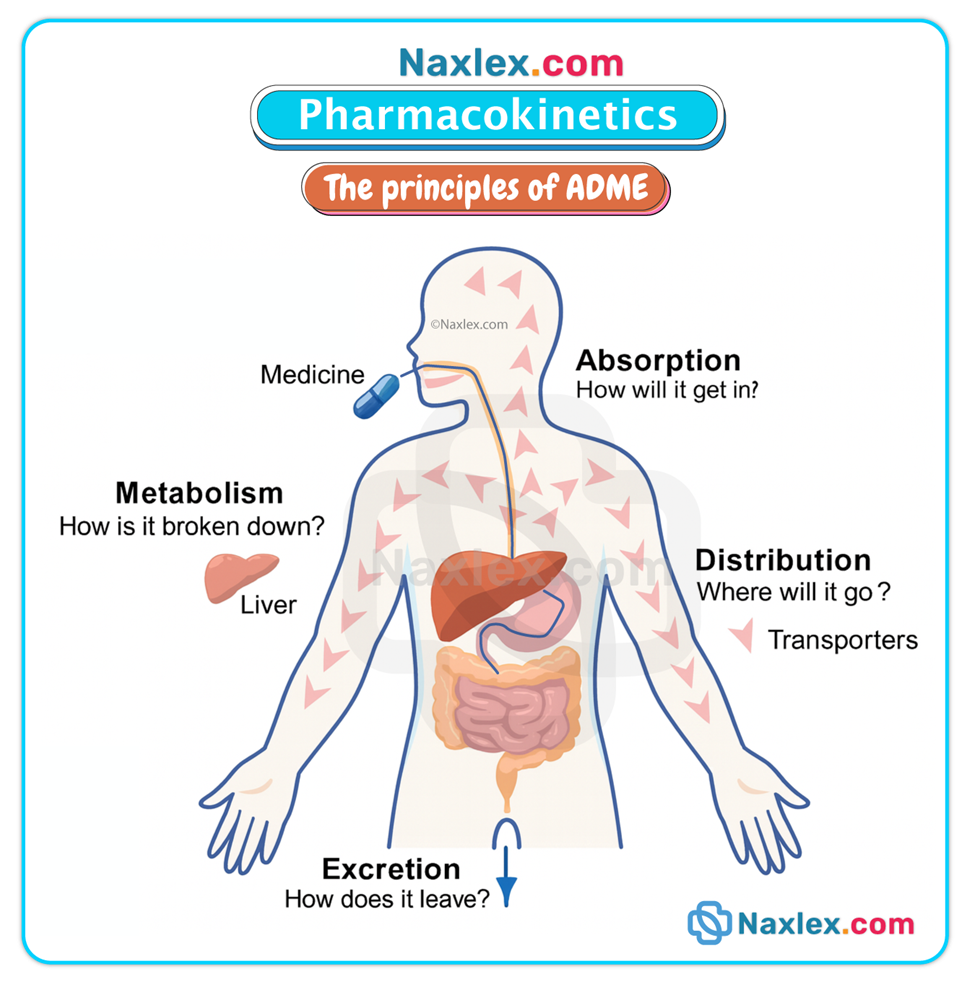
ABSORPTION
Definition & mechanisms
• Absorption is the movement of drug from the site of administration into the bloodstream (systemic circulation). Mechanisms include:
- Passive diffusion (most common; drug moves down a concentration gradient without energy),
- Active transport (requires carrier proteins and energy to move against gradients),
- Pinocytosis (cellular engulfment of drug particles).
Primary absorption sites & routes
• Oral drugs: primarily absorbed in the small intestine across mucosal villi (large surface area).
• Parenteral: IV (direct into bloodstream; 100% bioavailability), IM and subcutaneous (absorption rate depends on blood flow and tissue vascularity).
• Other: sublingual, transdermal, inhaled, topical, rectal — each route has unique absorption kinetics and bypasses or partially bypasses first-pass liver metabolism.
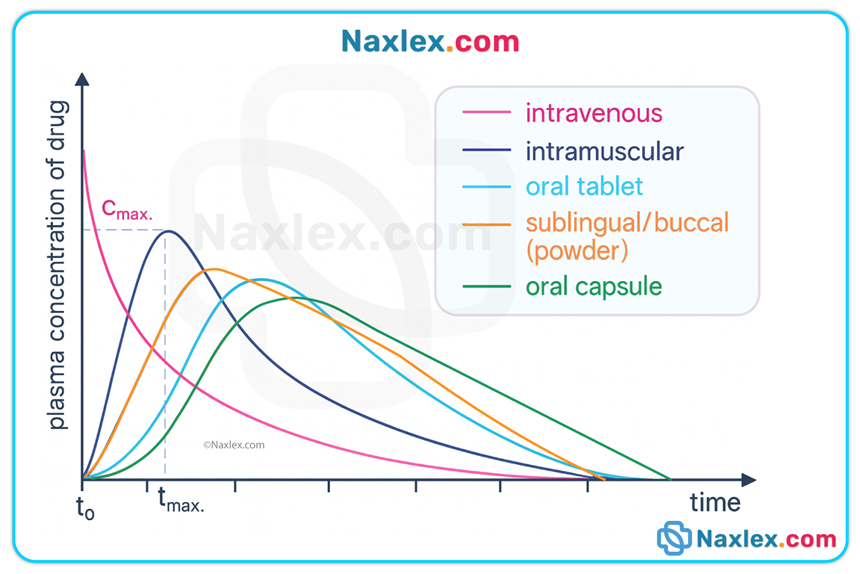
Factors influencing absorption
• Drug properties: lipid solubility (lipid-soluble drugs cross membranes faster), particle size, ionization (nonionized are absorbed more readily).
• pH & ionization: acidic drugs are less ionized in acidic environments (e.g., aspirin in stomach), facilitating absorption there; basic drugs absorb better in more alkaline environments.
• Blood flow: perfusion at the absorption site (e.g., shock, vasoconstriction, local edema) decreases absorption.
• Surface area & integrity: loss of mucosal villi (celiac disease, extensive resections) or mucosal disease reduces absorption.
• First-pass effect: after absorption from the GI tract, drugs reach the liver via portal circulation and may be partially metabolized before reaching systemic circulation, reducing bioavailability. Examples of drugs with extensive first-pass metabolism include certain nitrates and many analgesics; such drugs may require higher oral doses or alternate routes.
• Co-administration factors: food, other drugs (antacids, enzyme inducers/inhibitors), exercise, and physiologic states (fasting, stress) alter absorption.
Clinical & pathophysiologic links
• Infants: higher gastric pH → greater absorption of acid-labile drugs (e.g., increased penicillin absorption).
• Older adults: reduced GI motility and lower gastric acid can slow absorption of some drugs.
• Shock/hypoperfusion: reduced splanchnic blood flow delays oral absorption and may shift reliance to parenteral routes.
• IM injection site: muscles with greater blood flow (deltoid) absorb faster than less vascular sites (gluteus).
Bioavailability
• Bioavailability = percentage of an administered dose that reaches systemic circulation as active drug. IV = 100%; oral bioavailability is typically <100% due to incomplete absorption and first-pass metabolism. Factors altering bioavailability: formulation, route, GI mucosa/motility, presence of food/other drugs, hepatic function.
Nursing insights
• Select route appropriate to clinical condition (e.g., avoid oral if rapid effect needed or GI absorption impaired).
• Follow administration instructions regarding food and fluids, and educate patients.
• Monitor for delayed or reduced effect in conditions that alter perfusion or GI integrity.
• Recognize that co-prescribed drugs (antacids, PPIs, enzyme inducers/inhibitors) may alter absorption and bioavailability.
DISTRIBUTION
Definition & core determinants
• Distribution refers to the reversible transfer of a drug between the bloodstream and tissues. Key determinants include blood flow to tissues, capillary permeability, tissue affinity for the drug, and plasma protein binding.
Volume of distribution (Vd)
• Vd is a theoretical concept describing how a drug distributes between plasma and the rest of the body; drugs with large Vd often reside in tissues and may have prolonged half-lives.
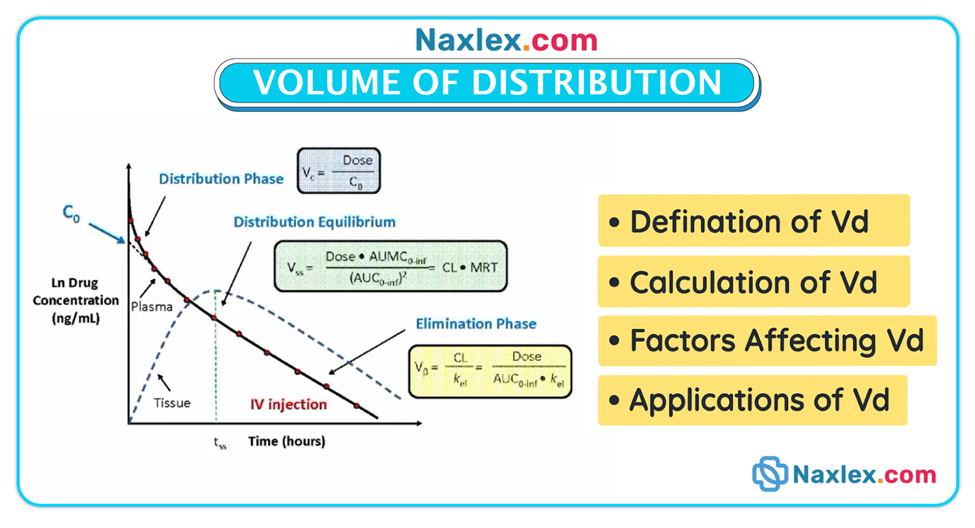
Protein binding
• Many drugs bind to plasma proteins (primarily albumin). Bound drug is pharmacologically inactive; only the free (unbound) fraction is available to cross membranes, interact with receptors, and be eliminated.
• Ranges (common classification used clinically):
- High protein binding: >89% bound
- Moderately high: 61–89%
- Moderate: 30–60%
- Low: <30%
• Clinical consequences: When two highly protein-bound drugs are coadministered, they compete for binding sites → increased free drug fraction → risk of toxicity. Low albumin states (malnutrition, liver disease, burns, nephrotic syndrome) decrease binding capacity → increased free drug → potential toxicity.
Barriers & tissue sequestration
• Blood–brain barrier (BBB): tight junctions and lipid-rich cell membranes restrict passage; lipid-soluble, unbound drugs cross most readily. Inflammatory states (meningitis) can disrupt BBB and allow entry of normally excluded drugs.
• Placental barrier: many drugs cross and can affect the fetus; both lipid-soluble and some water-soluble drugs cross the placenta—risk-benefit must be carefully weighed in pregnancy.
• Breast milk: drugs may be secreted and affect nursing infants; knowledge of lactation compatibility is necessary.
• Tissue accumulation: some drugs accumulate in fat (lipophilic drugs), bone, liver, muscle, or ocular tissues, which can create depots that prolong effect and complicate toxicity.
Distribution impediments
• Abscesses & exudates: minimal blood supply and acidic environment reduce antibiotic penetration; drainage and local management may be required to achieve therapeutic effect.
• Tumors and poorly perfused tissues may receive inadequate drug levels.
Nursing insights
• Assess nutritional status, albumin levels, and conditions affecting protein binding.
• Anticipate altered dosing in hypoalbuminemia and with polypharmacy involving highly protein-bound drugs.
• Consider tissue sequestration when planning duration and monitoring for toxicity.
• Be cautious with CNS-active drugs in patients with disrupted BBB.
• Review pregnancy and lactation status before prescribing potentially teratogenic or excretable drugs.
METABOLISM (BIOTRANSFORMATION)
Purpose & major site
• Metabolism converts drugs (often lipophilic) into more polar, water-soluble metabolites that can be readily excreted, predominantly by the liver using enzyme systems such as cytochrome P450 (CYP) families. Metabolism may inactivate a drug, produce an active metabolite, or convert prodrugs into active forms.
Phases of metabolism
• Phase I (functionalization reactions): oxidation, reduction, hydrolysis — frequently mediated by CYP enzymes.
• Phase II (conjugation reactions): glucuronidation, sulfation, acetylation — attach polar groups to increase water solubility.
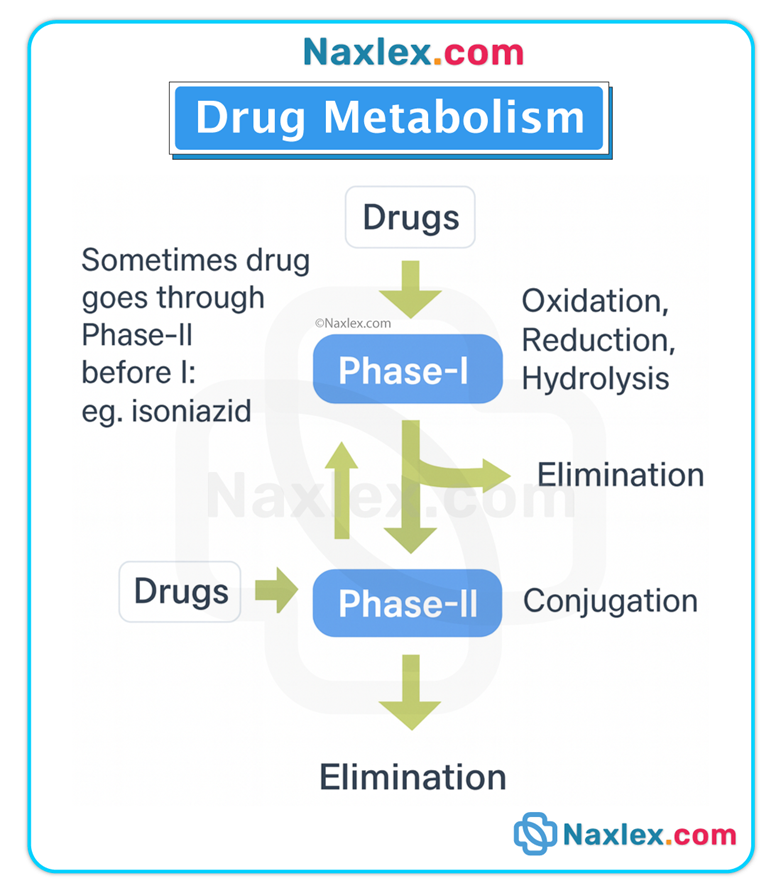
Genetic & physiologic variability
• Pharmacogenetics: genetic polymorphisms in metabolic enzymes produce fast or slow metabolizers for particular drugs (e.g., CYP2D6, CYP3A4 variants) and can dramatically affect drug levels and responses.
• Age: neonates/infants have immature hepatic enzyme systems; older adults often have reduced hepatic blood flow and enzyme activity.
• Disease: hepatic disease (cirrhosis, hepatitis) reduces metabolic capacity, prolongs drug half-life, and increases toxicity risk.
• Drug–drug interactions: enzyme inhibitors (decrease metabolism → increased levels/toxicity) and inducers (increase metabolism → decreased levels/therapeutic failure).
First-pass (hepatic) effect
• Drugs absorbed from the gastrointestinal tract pass through the portal circulation to the liver where a significant fraction may be metabolized before reaching systemic circulation. Drugs with high first-pass metabolism have reduced oral bioavailability — alternative routes (sublingual, transdermal, parenteral) or higher oral doses may be necessary.
Half-life (t½) and steady state
• Half-life is the time required for the drug concentration to fall by 50%. It reflects combined effects of metabolism and elimination. Short half-life (≈4–8 hours) means faster elimination and often more frequent dosing; long half-life (≥24 hours) means prolonged presence and less frequent dosing.
• Steady state is achieved after approximately 3 to 5 half-lives of consistent dosing; at steady state the rate of drug intake equals rate of elimination. Example: digoxin (half-life ≈36 hours in normal renal function) requires several days to reach steady state.
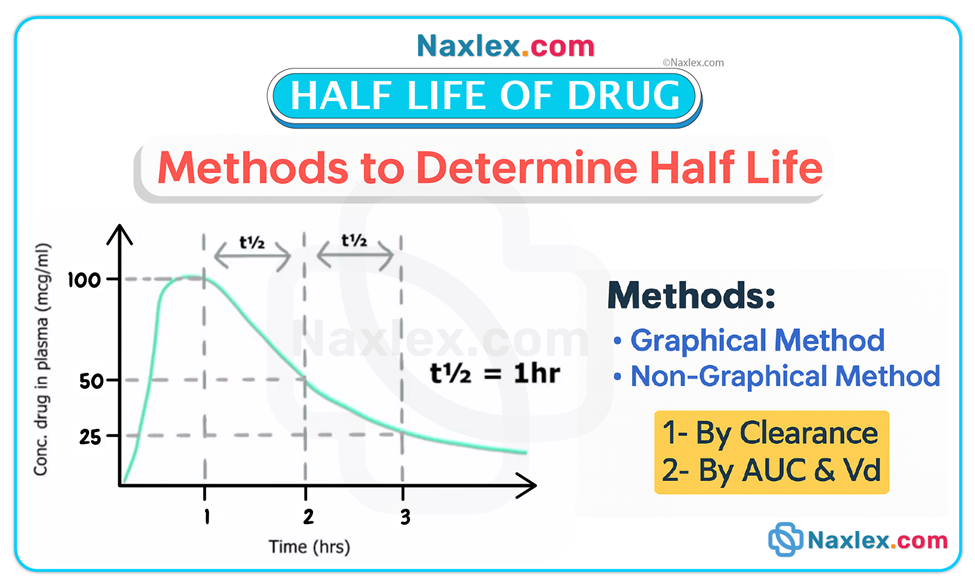
Active metabolites
• Some drugs produce metabolites that are equally or more active than the parent compound (e.g., certain morphine metabolites), altering the duration and nature of effect and sometimes increasing toxicity.
Nursing insights
• Review hepatic function tests (AST, ALT, total bilirubin) when prescribing drugs predominantly metabolized by the liver.
• Anticipate dose reductions or alternative therapies in hepatic impairment.
• Recognize potential enzyme-mediated drug–drug interactions and consult pharmacy when adding or removing medications.
• Consider genetic variability where clinically significant (e.g., codeine activation via CYP2D6).
EXCRETION (ELIMINATION)
Primary routes
• Renal elimination (urine) is the most common route: kidneys filter free, water-soluble drugs and metabolites; active tubular secretion and reabsorption further influence final excretion. Other routes include bile/feces, lungs (volatile agents), sweat, saliva, and breast milk.
Renal function & drug clearance
• Glomerular filtration rate (GFR) and creatinine clearance (CLcr) are key determinants of renal elimination. Renal impairment (acute or chronic) reduces clearance and increases the risk of drug accumulation and toxicity.
• Creatinine clearance is used to adjust dosing for renally excreted drugs; CLcr varies with age, gender, and muscle mass (lower in older adults and women).
Urine pH manipulation
• Urine pH alters tubular reabsorption: acidic urine promotes excretion of weak bases; alkaline urine promotes excretion of weak acids. Clinically, sodium bicarbonate may be used to alkalinize urine to enhance elimination of aspirin (salicylate) in overdose. Conversely, substances that acidify urine (e.g., large amounts of cranberry juice) could theoretically reduce excretion of weak acids.
Extra-renal elimination
• Biliary excretion: drugs excreted in bile may be reabsorbed via enterohepatic circulation, prolonging effect.
• Pulmonary excretion: volatile anesthetics are eliminated via exhalation.
Nursing insights
• Monitor BUN, serum creatinine, CLcr, and urine output to detect changes in elimination capacity.
• Adjust dosing intervals and/or doses for renal impairment; consult dosing guidelines for renally cleared drugs.
• Avoid or use caution with nephrotoxic drugs (aminoglycosides, certain antivirals, NSAIDs) in patients with renal dysfunction.
• Ensure adequate hydration when appropriate to maintain renal perfusion and elimination.
Pharmacodynamic Phase
(What the drug does to the body — mechanism and effect)
Definition & scope
• Pharmacodynamics concerns the biochemical and physiologic effects of drugs and the mechanisms by which those effects are produced, including receptor interactions, dose–response relationships, efficacy, potency, and the time course of pharmacologic effects.
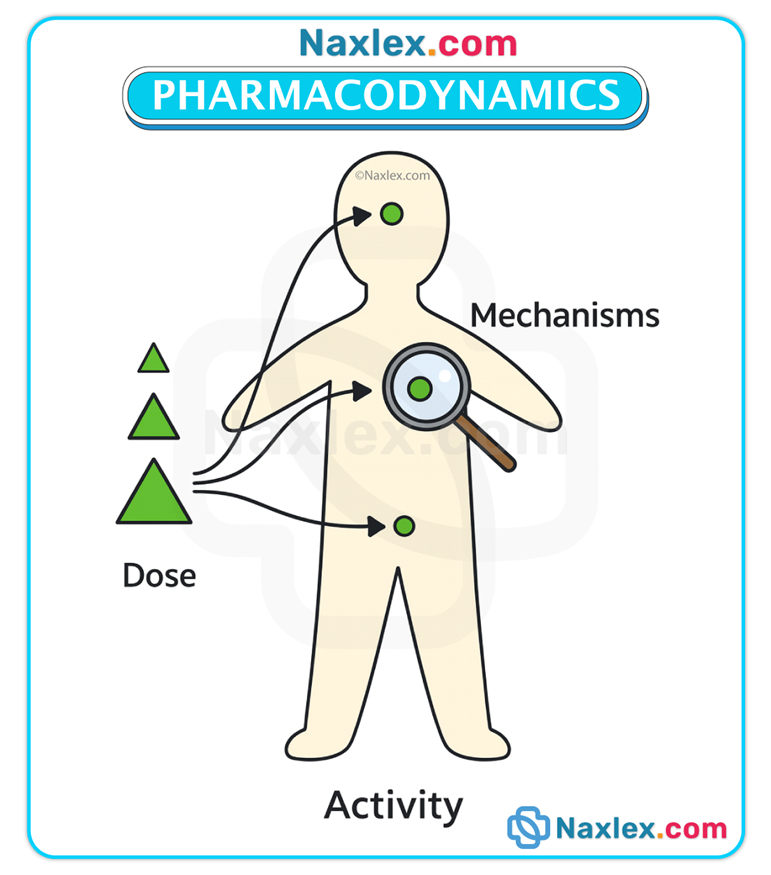
Primary vs secondary effects
• Primary effect: the therapeutic, intended response (e.g., antihypertensive lowering blood pressure).
• Secondary effect: any additional effect (beneficial or adverse) that occurs because of the drug’s mechanism (e.g., diphenhydramine — primary: antihistamine; secondary: sedation).
Dose–response & maximal efficacy
• Dose–response relationship: increasing dose generally increases effect up to a point. Potency refers to the amount of drug needed to produce a given effect; efficacy refers to the maximal effect achievable. Example: morphine has greater maximal analgesic efficacy than tramadol even if doses are increased.
Onset, peak, and duration
• Onset: time to reach the minimum effective concentration (MEC) after administration.
• Peak: time of highest plasma concentration (and often maximum effect).
• Duration: period during which the drug concentration remains above MEC. Understanding these guides timing of doses and monitoring.
Receptor theory and receptor families
• Drugs typically act by binding to receptors — protein structures on cell membranes or inside cells that mediate effects. The quality of fit between drug and receptor dictates magnitude of response. Major receptor families:
- Kinase-linked receptors: ligand binds extracellular domain, activates intracellular kinase → modifies cellular enzymes/transcription. Response typically intermediate in onset.
- Ligand-gated ion channels: binding opens ion channel (Na⁺, Ca²⁺) → very rapid responses (milliseconds to seconds).
- G protein–coupled receptors (GPCRs): receptor activates a G protein which then alters effectors (enzymes, ion channels) — common and versatile signaling mechanism.
- Nuclear receptors: located inside the cell nucleus; activation influences gene transcription and protein synthesis — effects are slower in onset but prolonged.
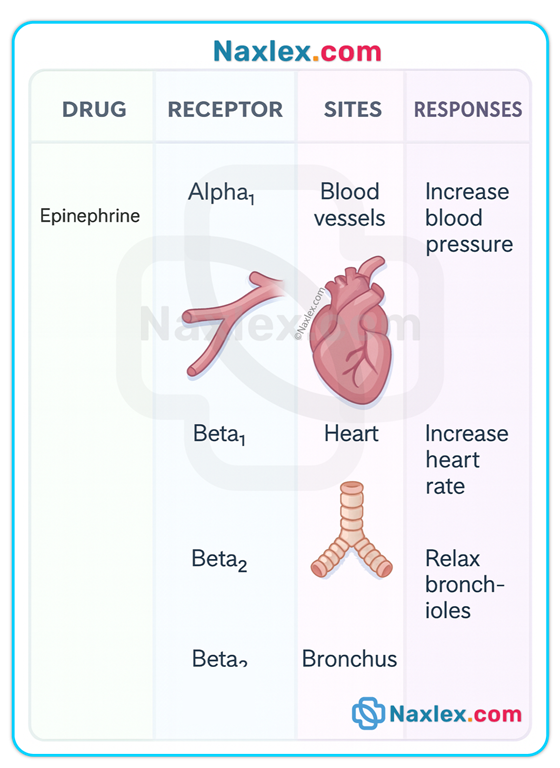
Agonists, antagonists, partial agonists
• Agonist: binds receptor and elicits full biologic response (e.g., epinephrine on beta receptors).
• Antagonist: binds receptor and blocks agonist action (no intrinsic activity). IC50 quantifies the antagonist concentration required to inhibit 50% of maximum response.
• Partial agonist: elicits less than maximal response even at full receptor occupancy.
Nonselective & nonspecific effects
• Nonspecific: drug acts on physical or chemical processes not limited to one receptor type (e.g., osmotic diuretics).
• Nonselective: drug affects multiple receptor subtypes leading to varied effects at different organs (e.g., bethanechol stimulates cholinergic receptors in bladder, heart, bronchi, and GI tract → bladder contraction + bradycardia + bronchoconstriction + increased gastric secretions).
Categories of drug action
- Stimulation or depression of cellular activity (e.g., stimulants vs sedatives).
- Replacement therapy (e.g., insulin replaces endogenous hormone).
- Inhibition or killing of organisms (antibiotics, antifungals).
- Irritation (laxatives that increase peristalsis by local irritation).
Therapeutic index & therapeutic range (window)
• Therapeutic index (TI) = LD50 / ED50. TI is a crude measure of safety: a small TI (close to 1) indicates narrow margin between effective and lethal doses; such drugs require careful monitoring.
• Therapeutic range = plasma concentrations between MEC and minimum toxic concentration (MTC). Some drugs (e.g., digoxin, aminoglycosides, anticonvulsants) have narrow therapeutic ranges and require periodic serum level checks.
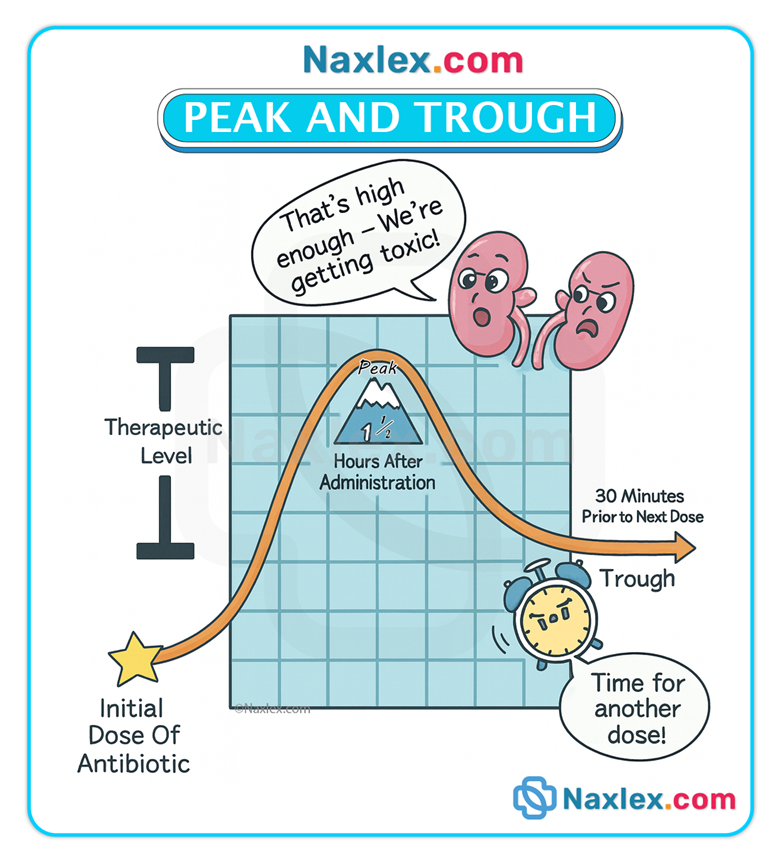
Peak & trough monitoring
• Peak level: indicates rate/degree of absorption; drawn at time of expected peak (varies by route: e.g., 1–3 hours post-oral or ~10 min after IV bolus for many drugs).
• Trough level: indicates elimination; drawn immediately before the next dose.
• Monitoring prevents underdosing (low peak) and toxicity (high trough).
Loading dose & digitalization
• A loading dose is an initial large dose given to rapidly achieve therapeutic plasma concentration; subsequent maintenance doses sustain that level. Digoxin digitalization is a classic example where loading doses achieve therapeutic steady state more quickly.
Side effects, adverse reactions, and toxicity
• Side effects: expected, often predictable physiologic effects related to mechanism (may be harmless or bothersome).
• Adverse reactions: more severe, unintended, and undesirable effects occurring at normal doses (e.g., anaphylaxis). Always report and document.
• Toxicity: excessive drug levels produce harmful effects (often preventable with monitoring). Management may require dose reduction, antidotes, or enhanced elimination.
Pharmacogenetics
• Genetic variations alter pharmacokinetics and pharmacodynamics (e.g., metabolism rates differ by CYP genotype), leading to variable drug responses and risk of adverse effects. Ethnic and genetic population differences may influence drug selection and dosing; individual genotyping is increasingly relevant for certain drugs.
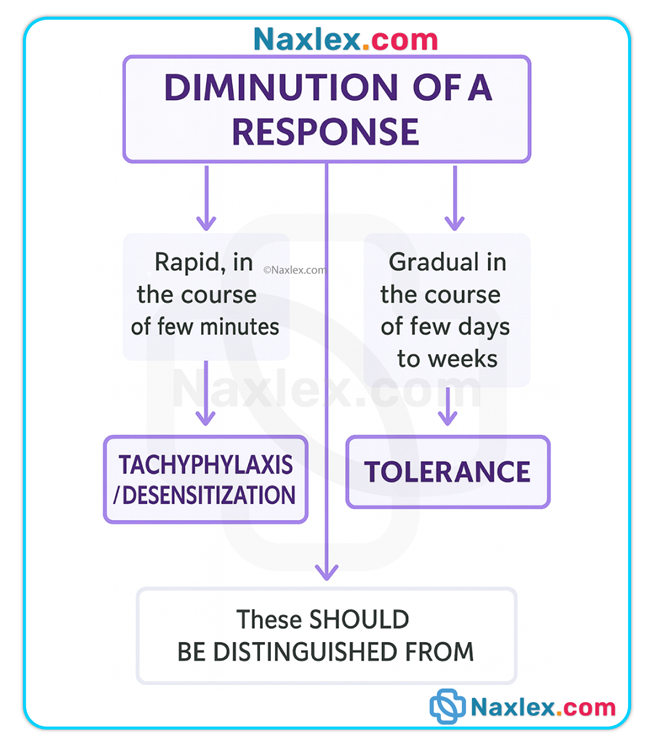
Tolerance & tachyphylaxis
• Tolerance: gradual decrease in response over time requiring higher doses for same effect (seen with opioids).
• Tachyphylaxis: rapid development of decreased responsiveness (acute tolerance), seen with some sympathomimetics, nitrates, and psychotropic agents.
Placebo effect & ethical considerations
• Placebo responses are real psychophysiologic improvements due to expectation rather than pharmacologic action; their use is limited by ethical obligations to informed consent in clinical practice.
Nursing insights
• Know whether a drug is an agonist, antagonist, or partial agonist and the clinical implications of each.
• Monitor for expected therapeutic effect and for known class-specific adverse effects.
• Understand drug onset/peak/duration to coordinate assessments, safety precautions (e.g., fall risk when peak sedation expected), and patient education.
• Obtain baseline and periodic laboratory monitoring when indicated (serum drug levels, LFTs, renal function).
• Be aware of genetic and physiologic patient factors that may alter response.
Additional Concepts And Clinical Applications
Half-life and steady-state application
• Use half-life to anticipate accumulation, duration, and time to steady state. Multiple half-lives are required to eliminate a drug (>90% elimination after about 4–6 half-lives). Adjust dosing frequency based on half-life to avoid accumulation.
Drug interactions — protein binding and metabolism
• Protein displacement: two highly protein-bound drugs coadministered may increase free concentrations of one or both agents → toxicity.
• Metabolic interactions: enzyme inhibitors (e.g., some antifungals) and inducers (some anticonvulsants) change plasma drug levels — monitor closely and adjust dosing.
Special populations
• Pediatrics: immature hepatic enzymes, higher body water proportion, and higher gastric pH alter pharmacokinetics; dosing often weight-based and requires age-specific considerations.
• Older adults: decreased renal function, reduced hepatic blood flow, altered body composition (higher fat:lean ratio, lower albumin) → altered distribution, metabolism, and excretion; start low and go slow when dosing.
• Pregnancy & lactation: evaluate teratogenic risk, placental transfer, and excretion into breast milk; weigh maternal benefit vs fetal/neonatal risk.
Routes that bypass the pharmaceutic phase
• Parenteral (IV, IM, subQ), transdermal, sublingual, inhalational, ophthalmic, and intranasal routes bypass or significantly alter the pharmaceutic phase and have distinct absorption profiles and monitoring needs.
Practical nursing actions for safe medication use
• Always verify route and formulation before administration.
• Know which medications must not be crushed.
• Check compatibility for IV mixtures and infusion rates.
• Confirm allergy history and document adverse reactions.
• Monitor therapeutic and adverse effects and appropriate labs (drug levels, renal and hepatic function).
• Educate patients on timing relative to meals, expected onset, and warning signs of toxicity or adverse effects.
• Report and document adverse drug events per facility policy.
Summary
Three phases of drug action:
- Pharmaceutic (oral solids only — disintegration/dissolution)
- Pharmacokinetic (ADME)
- Pharmacodynamic (drug effect and mechanism).

• Pharmaceutic phase: affected by formulation (enteric-coated, sustained-release), excipients, gastric pH, motility, and presence of food. Do not crush enteric-coated or extended-release forms.
• Absorption: route, blood flow, lipid solubility, ionization, surface area, and first-pass hepatic metabolism influence how much and how fast a drug reaches systemic circulation. Bioavailability varies by route and formulation.
• Distribution: blood flow, tissue affinity, protein binding, and barriers (BBB, placenta) determine drug movement to site of action. Hypoalbuminemia increases free drug and toxicity risk. Volume of distribution influences half-life.
• Metabolism: primarily hepatic via enzyme systems; can inactivate drugs or produce active metabolites. Genetic differences and liver disease greatly affect metabolism. Half-life and steady state are functions of metabolism/elimination.
• Excretion: kidneys are the primary route for most drugs/metabolites; renal impairment necessitates dose adjustment. Urine pH can be manipulated to enhance elimination of certain drugs.
• Pharmacodynamics: drugs act via receptors, enzymes, ion channels, and other mechanisms; potency and efficacy differ between agents. Therapeutic index and therapeutic range determine monitoring needs.
• Monitoring & safety: peak/trough levels for narrow therapeutic index drugs, LFTs/renal tests for drugs cleared by those organs, vigilance for adverse reactions and interactions.
• Clinical implications: age, pregnancy, organ dysfunction, genetic variability, and polypharmacy require individualized dosing, close monitoring, and patient education.
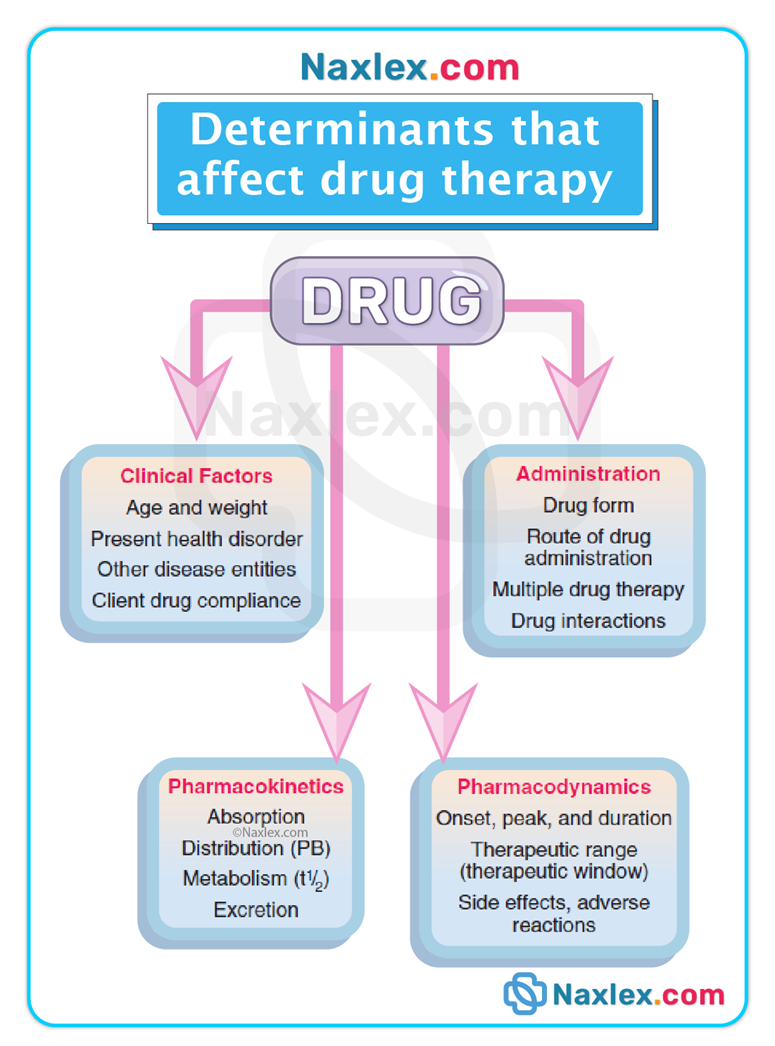
Nursing Insights:
• Verify formulation before administration (do not crush SR/ER/enteric-coated).
• Confirm route appropriateness for clinical status (IV when rapid effect required or absorption impaired).
• Assess hepatic and renal function and adjust dose as indicated; obtain CLcr when dosing renally excreted drugs.
• Monitor serum drug levels where applicable (aminoglycosides, anticonvulsants, digoxin).
• Evaluate for drug interactions (protein binding displacement, enzyme induction/inhibition).
• Educate patients about administration timing with food, potential side effects, and signs of toxicity.
• Consult pharmacy for complex dosing decisions, interactions, and alternative formulations.
Naxlex
Videos
Login to View Video
Click here to loginTake Notes on Drug action: pharmaceutic, pharmacokinetic, and pharmacodynamic phases
This filled cannot be empty
Join Naxlex Nursing for nursing questions & guides! Sign Up Now
